
Seven points on the current state of AI in genomics
A grounded take from the messy middle of an AI revolution

Last Friday, the Committee on Genetics, Genomics & Systems Biology hosted its annual symposium at the University of Chicago, this year on the theme of AI in Genomics. We brought together six speakers whose work spans much of the interesting territory in the field right now: Katie Pollard (Gladstone/UCSF), Andrew Kern (Oregon), Alex Lu (Microsoft Research), Arjun Krishnan (Colorado), and our own Siwei Chen and Aly Khan from UChicago.

It was a full and packed day (six talks, a panel, a reception, and a dinner) and rather than try to recap each talk individually, I wanted to use the occasion to lay out what I think are the most interesting points about where AI in genomics stands right now. Some of these came directly from the talks; others are observations the day brought into focus.
1. DNA models have grown enormously - but haven’t yielded much biological insight yet
The progression of DNA models since 2021 has been dominated by scale. DNABERT (Ji et al., 2021) established the BERT-style self-supervised pretraining paradigm for DNA, which DNABERT-2 (Zhou et al., 2023) refined with byte-pair encoding and multi-species training; the Nucleotide Transformer family (Dalla-Torre et al., 2024) extended it across hundreds of species; Enformer (Avsec et al., 2021) and HyenaDNA (Nguyen et al., 2023) reached for longer genomic context. Then, earlier this year, Evo 2 (Brixi et al., 2026) jumped to a different scale entirely: 40 billion parameters, 9.3 trillion nucleotides spanning every domain of life, and a 1-million-token context window at single-nucleotide resolution.
The more uncomfortable observation is that this enormous scale-up has not translated into the kind of insight one might expect. And that’s because - according to Alex Lu - DNA is inherently different from natural language in several important ways: low signal-to-noise, vast tracts of repetitive sequence, no obvious analog to “words” or “sentences”, and functional elements that are sparse, distant, and combinatorial.
A clear illustration of this disconnect comes from Vishniakov et al. (2025). The authors evaluated seven genomic foundation models across 52 tasks and compared each pretrained model to randomly-initialized baselines of matched architecture. Their finding was striking: randomly initialized models often matched or even surpassed pretrained models in fine-tuning and feature extraction, and the pretrained models systematically failed to capture clinically relevant variants. Tang et al. (2025), evaluating pretrained DNA language model representations against raw one-hot encoded sequences across six regulatory genomics tasks, reached compatible conclusions: the learned representations offer little to no advantage over the raw sequence itself.
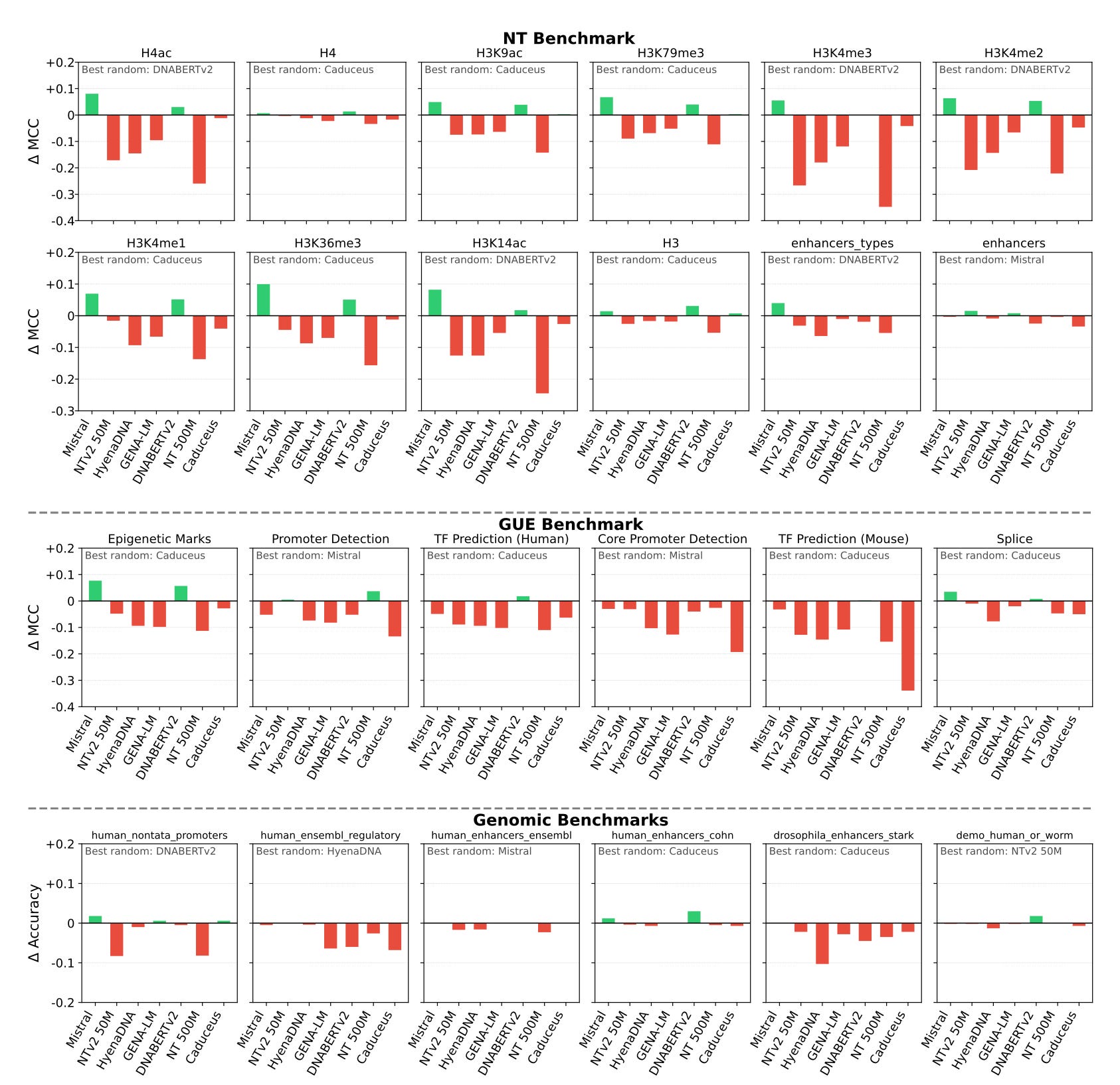
The lesson is that scale alone does not deliver biological understanding. Which leads to my next point:
2. Informed models are key
If brute-force pretraining does not reliably produce useful representations, what does? A recurring theme across the symposium (explicit in some talks, implicit in others) was that ignoring theory and history is a mistake.
Genomics is not a young field. As Andy Kern pointed out, population and quantitative genetics have 100+ years of theory to draw on, beginning with the foundational work of Fisher, Wright, and Haldane on the modern synthesis. Similarly, bioinformatics, evolutionary genomics, and comparative genomics have decades of accumulated approaches and intuitions, including the empirical demonstration that functionally important non-coding regions are evolutionarily conserved across distant species (Hardison, 2000; Siepel et al., 2005). AI in genomics that ignores this body of knowledge is essentially trying to rediscover, from data alone, things we already know.
Alex Lu’s work is a clean example of doing this right. In his “reverse homology” approach (Lu et al., 2022), he used evolutionary conservation as the contrastive signal for a deep learning model, leveraging the fundamental principle from comparative genomics that functionally important sequences are conserved over evolution. Rather than trying to learn what matters from scratch, the model is told what sequences are likely important. The result was a small, interpretable model that successfully identified meaningful features of intrinsically disordered regions, something the larger, uninformed approaches had struggled with. Recent follow-up work building on this conservation-as-pretraining-signal idea (Karollus et al., 2024) supports the broader principle.
The general lesson: small, informed models can outperform large, uninformed ones. Leveraging 100 years of theory and prior biological knowledge, rather than asking gradient descent to recover it from scratch, is a much more efficient research strategy.
3. Predictions are fast; validation is slow
A point Katie Pollard made forcefully in her talk, and one that I think the broader AI-in-genomics community underappreciates, is the asymmetry between how fast we can generate predictions and how slowly we can validate them.
Modern models can score every variant in the human genome for predicted effect in a matter of hours. Generating a ranked list of candidate causal variants, predicted regulatory elements, or candidate disease genes is now cheap. But validating any single one of those predictions (e.g. establishing that a specific non-coding variant actually drives a specific phenotype through a specific mechanism) typically requires generating a mouse model, running cell-based functional assays, or executing CRISPR perturbation screens, each of which takes months to years and substantial cost. Pollard’s recent work illustrates exactly this dynamic. Her group developed CardioAkita, a machine-learning model that predicts how structural variants alter 3D chromatin organization, and used it to score de novo structural variants from individuals with congenital heart disease. The computational predictions identified disruptive variants that were then validated experimentally: induced pluripotent stem cells engineered to carry the predicted variants showed the predicted 3D chromatin changes and aberrant expression of cardiac developmental genes (Lee et al., 2026). The computational step was fast; the experimental validation took years.
This asymmetry has a clear implication: the highest-leverage methodological investments right now may not be in better models, but in high-throughput, fast validation platforms that can keep up with computational output. Approaches like massively parallel reporter assays (MPRAs), Perturb-seq, base-editor screens, saturation mutagenesis, and iPSC-engineered variant panels are critical, because they turn AI-prioritized hypotheses into testable, validated biology at scale. This is not a new framing; this has been explicit in protein engineering and synthetic biology for years, where the Design-Build-Test-Learn cycle treats closing the experimental loop as a research frontier in its own right (Carbonell et al., 2018). Genomics is now converging at the same realization.
4. Vast genomic data exists — the bottleneck is metadata
Arjun Krishnan made the extraordinary estimate in his talk that there are roughly 2.5 million human transcriptomes publicly available across resources like SRA and GEO. That is an incredible resource that every other field would die for, and the potential AI applications are staggering. But it is hugely underutilized.
The catch is that the metadata are non-uniform, often incomplete, frequently wrong, and usually unstructured. Tissue annotations vary across studies, disease labels are inconsistent, sample preparation and sequencing details are missing. So the data are there, but without high quality metadata, it is challenging to extract biological signal at scale. This problem has been documented across multiple efforts to harmonize public expression data (Bernstein et al., 2017). This is not unique to RNA-seq, and consistently found across any type of public data, including microbiome.
This means that some of the highest-leverage methodological work in the field right now is not in modeling, but in building tools that improve, harmonize, and validate metadata. It is unglamorous, but it is where a huge fraction of the value will come from.
5. Be skeptical of all benchmarking
Every new model is the best on its own benchmarks. This is not a controversial statement; it is an observation about the literature, and one that has been documented systematically in adjacent fields. Whoever publishes a model also typically chooses the benchmark, and the benchmark almost invariably flatters the model. Even where benchmarks are shared, evaluation protocols, hyperparameter budgets, and dataset splits often differ in ways that advantage the proposing method.
Arjun Krishnan shared a rule of thumb that I think is genuinely useful: the best model is usually the one that is consistently number 2 in benchmarks across the literature. This rule has straightforward reasoning: a model that is consistently competitive but rarely the absolute winner is more likely to be genuinely strong than one that is ranked #1 only on the benchmark its authors designed. I think this is a practical heuristic that reflects how publication incentives shape benchmark design.
6. AI tools are disruptive for trainees, but a real boon when used well
A question that came up repeatedly, both in the panel and in side conversations during breaks, was how graduate students should think about AI tools. There is a clear tension: on one hand, code-generation assistants and large language models can dramatically increase productivity. On the other hand, leaning on these tools too heavily can hollow out the deep understanding a PhD is supposed to build.
A useful rule of thumb is this: if you can validate what the AI produces – namely, if you can do the task yourself comfortably and check whether the AI did it correctly – then you can probably use AI to do the task. Otherwise, you should probably do it yourself, even if it feels hard. The friction of doing something the slow way is often the friction of actually learning it, and an AI tool that produces output you cannot evaluate is a black box you are forced to trust.
This is also a place where PIs need to take the lead. Writing down explicit AI use guidelines for your lab is more straightforward than it might seem, and I have written about how to do it here.
You need to make AI guidelines for your lab
·Generative AI is now part of academic research. A recent Nature feature found that PhD students are using AI tools daily to search the literature, write code, draft manuscripts, analyze data, and interpret results (Nordling 2026). Some use it constantly and swear by it; others avoid it, worried about the cost to their development. Most fall somewhere in…

7. There is a backlash against AI in academia, and we should be careful not to lump everything together
A recurring undercurrent at the symposium, and one I think is worth naming, is that academic culture has begun to develop a real skepticism toward AI, sometimes even a knee-jerk hostility. Some of this is understandable: there are real concerns about training data provenance, environmental costs (Strubell et al., 2019), the displacement of human expertise, the use of AI tools by students to bypass coursework, and the flood of low-quality AI-generated content in the literature (Liang et al., 2024).
But a lot of this hostility conflates very different things. When most people say “AI,” they are thinking about ChatGPT, a general-purpose chatbot whose flaws and risks are well-documented. The work the symposium speakers presented is something else entirely: domain-specific models, built and evaluated by careful scientists, used as one tool among many to make progress on hard biological problems. Treating these as the same thing, and rejecting both wholesale, is not productive.
The position I support is the one our speakers occupy, and it is more interesting than wholesale enthusiasm or wholesale rejection of AI: as scientists, we should engage seriously with specific claims, on specific tasks, with specific evidence. The general lesson (which matters beyond AI) is that a defensible opinion on a complex topic is rarely a single sentence. It has to be built case by case, grounded in actual understanding, and updated as the evidence evolves. Almost nothing is one-sided, despite how it often appears from social media or polarized public discourse. The work of building that kind of grounded, case-by-case judgment is not easy, but it is one of the most valuable habits academic training can develop.
Closing
The symposium reinforced something I have come to believe more strongly over the past year: AI in genomics is moving past its first hype cycle. The questions are becoming sharper: when AI helps, which methods actually generate biological insight, how to integrate these tools with the field’s existing theoretical foundations and experimental platforms. And the answers will require deeper engagement with biology, not bigger models.
For the genomics community, and especially our trainees, this is a remarkably exciting moment.
Great post. I wrote about point 3 here - IMO it's helpful to think of validation as one thing, but curated to the class of answers we care about: https://norngroup.substack.com/p/ask-not-what-ai-can-do-for-longevity